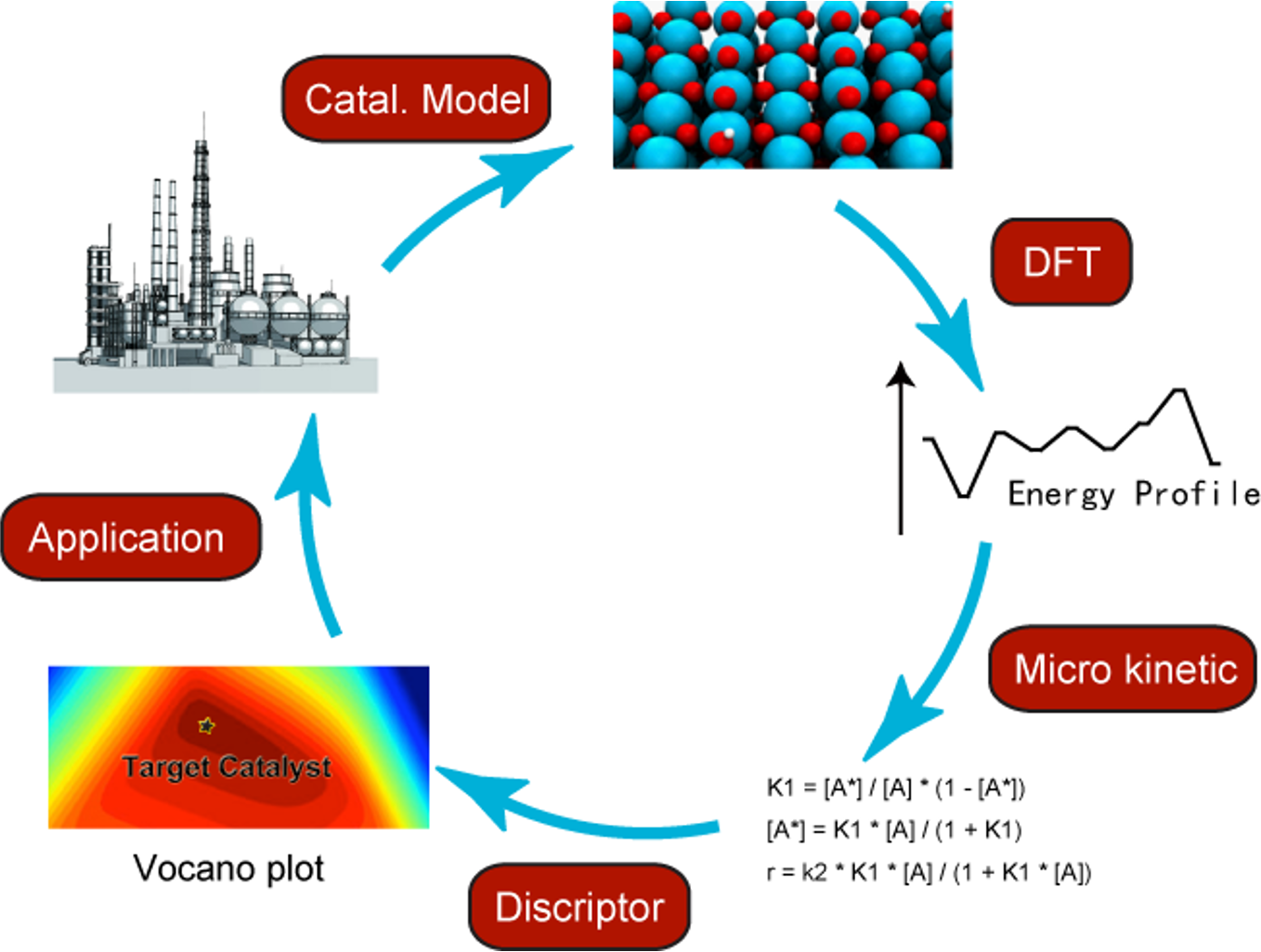
AI + Catalysis
Exploration of catalyst reaction mechanisms is of fundamental importance in improving known catalysts or designing new catalysts. In recent years, a great number of the catalytic processes have been computationally studied using Density Functional Theory (DFT) calculations and other relative methods. Along with the development of computational chemistry methods, parallel computing and high-performance computing cluster, state-of-the-art computational chemistry researches not only uncover the essence of a known catalytic process, but also are used as a fast and low-cost pre-screening technic to assist new catalyst design.Currently, we are focusing on following projects
1. Mechanistic research on propane dehydrogenation to propylene
 In order to gain deeper understanding of dehydrogenations of saturated hydrocarbons, we selected propane dehydrogenation reactions as our model reactions, which was catalyzed by Pt based catalyst. To facilitate determination of systematic trends in the propane dehydrogenationon Pt based catalyst, we will first investigate propane dehydrogenation on flat and stepped Pt and PtM alloy surfaces as well as interfaces of metal oxides, including MgO, Al2O3, and TiO2. They were selected for initial study as supports with at least one surface which matches the shape and size of the Pt(100) or Pt(111) surface. In a practical sense, the matched lattice dimensions provide a more uniform geometry between systems, thus facilitating initial development of correlations and Brønsted–Evans–Polanyi (BEP) relationships at the metal/support interfaces. By systematically permuting the alloy metal and nature of the substrates, we will establish correlations and reactivity patterns, including the development of BEP relationships. In the future, the analysis can be extended to other supported transition metal catalysts, and "volcano" relationships can be constructed between the predicted activity of different metal alloy/support structures and key catalytic parameters that are identified through the analysis.
In order to gain deeper understanding of dehydrogenations of saturated hydrocarbons, we selected propane dehydrogenation reactions as our model reactions, which was catalyzed by Pt based catalyst. To facilitate determination of systematic trends in the propane dehydrogenationon Pt based catalyst, we will first investigate propane dehydrogenation on flat and stepped Pt and PtM alloy surfaces as well as interfaces of metal oxides, including MgO, Al2O3, and TiO2. They were selected for initial study as supports with at least one surface which matches the shape and size of the Pt(100) or Pt(111) surface. In a practical sense, the matched lattice dimensions provide a more uniform geometry between systems, thus facilitating initial development of correlations and Brønsted–Evans–Polanyi (BEP) relationships at the metal/support interfaces. By systematically permuting the alloy metal and nature of the substrates, we will establish correlations and reactivity patterns, including the development of BEP relationships. In the future, the analysis can be extended to other supported transition metal catalysts, and "volcano" relationships can be constructed between the predicted activity of different metal alloy/support structures and key catalytic parameters that are identified through the analysis.
2. Development of Global Optimization Algorithm
One challenge to build a realistic catalyst model is to determining its most stable structure (global minimum) or set of lowest energy structures of a catalyst under reaction conditions. Although local minimum optimization technics have already been well developed, it could not guarantee the optimized structure to be the most stable one in case with complicated environment, like high adsorbate coverage, surface reconstruction, etc. Ideally, the global minimum can be located by exploring the whole potential energy surface with conducting numbers of local optimizations. We are interested in improvement of global optimization efficiency by reducing the number of local optimizations to obtain the desired global minimum with advanced computational algorithms like genetic algorithm, machine learning.

3. Machine learning potentials
Recently, the data-driven ML technique is emerging as a useful tool and surrogate model to accelerate the time-consuming simulation. Machine learning potentials (MLP), which directly learns the potential surface from ab initio calculations, have been developed to act as an energy calculator with high accuracy while maintaining the large speedup. Using MLP as a surrogate model, not only the thermodynamics for active sites, the evaluation of kinetic properties like reaction rate can also be accelerated. MLP can significantly extend the spatial and time scale of atomistic simulations, providing more opportunities to simulate large-scale catalyst systems while maintaining high accuracy. Besides, MLP can accelerate enhanced sampling simulations to predict the long-time-scale surface reaction using ab initio calculation. In our group, we tend to develop and apply MLP to accelerate the time-consuming, large-scale, and long-term simulation of catalytic system.